Angelmani sündroom (AS) iseloomustab füüsilise ja vaimse arengu viivitus. Angelmani sündroomiga inimesed vajavad pidevat elukestvat hooldust, kuna nad ei suuda enda eest hoolitseda ega ohte õigesti hinnata. Haruldane geneetiline haigus sai oma nime Briti lastearst Harry Angelmanilt, kes kirjeldas seda haigust esimesena teaduslikust aspektist 1965. aastal.
Mis on Angelmani sündroom?

© fancytapis - stock.adobe.com
Väliselt on Angelmani sündroomiga inimestel pea lame selg, väljaulatuva alalõuaga suur suu ja laia jalaga, koordineerimata kõnnak.
Angelmani sündroomi muud omadused on halvasti pigmenteerunud nahk ning väikesed käed ja jalad. Skolioos, selgroo S-kujuline kumerus, esineb sageli puberteedieas. 90 protsenti Angelmani sündroomiga inimestest põeb epilepsiat. Käitumise eripära väljendub muu hulgas rõõmsameelses meeleolus, kus esinevad sagedased naeratused ja naer, märgatav entusiasm vee vastu ja intensiivne füüsilise läheduse otsing.
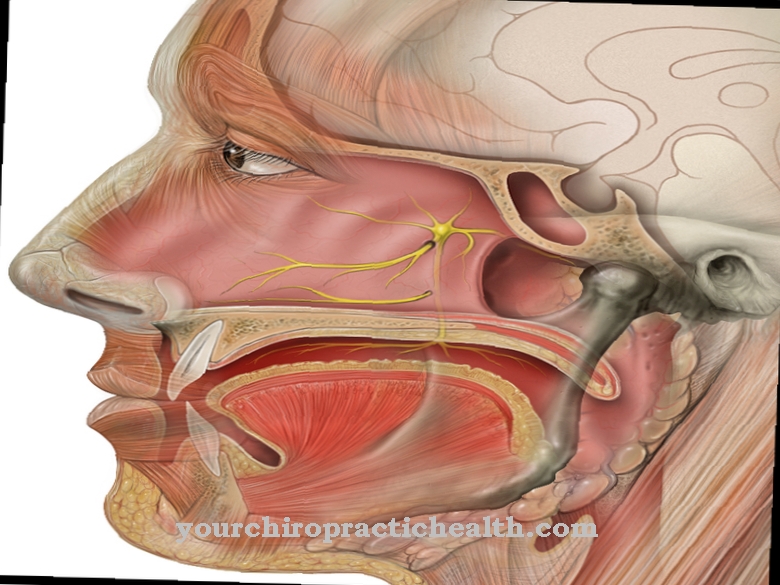
Samuti on Angelmani sündroomile iseloomulik keskendumisraskused, võimetus rääkida ja suukaudse faasi keskmisest suurem pikkus.
põhjused
Angelmani sündroomi põhjused on pärilikud. Kõige tavalisem on emalt päritud kõrvalekalle 15. kromosoomis. Kõige sagedamini põhjustab see kõrvalekalle kromosoomi väikeste lõikude puudumist.
Seevastu kümme protsenti anomaaliast on geenimutatsioon. Nendel juhtudel ei leitud Angelmani sündroomiga lapse emal defektseid lõike 15. kromosoomis. Seetõttu on võimalik, et kõrvalekalded tekkisid munaraku arengu ajal.
Üksikjuhtudel võib Angelmani sündroomi põhjuseks olla ka see, et mõlemad kromosoomid 15 pärinevad üksi isalt. Umbes kümnel protsendil Angelmani sündroomiga inimestest ei olnud 15. kromosoomi paaris kõrvalekaldeid. Geneetilise meigi muutus võib olla liiga väike ja seda ei saa praegu saadaoleva tehnoloogia abil kindlaks teha.
Sümptomid, tervisehäired ja nähud
Angelmani sündroom avaldub mitmesuguste füüsiliste sümptomite ja käitumise kaudu. Tüüpilised nähud ilmuvad tavaliselt alles pärast 3. eluaastat ja mõjutavad kogu keha. Esiteks kannatavad lapsed liikumis- või tasakaaluprobleemide all, mille raskusaste võib olla erinev. Tüüpilised on käte ja jalgade koordineerimata kõnnak ning tõmblevad liigutused.
Lisaks on võimalik kindlaks teha hüpermotoorne ja hüperaktiivne käitumine - mõjutatud on rahutud ja liiguvad katkematult. Keele areng on tavaliselt halvenenud. Mõjutatud isikud saavad aru, mida neile öeldakse, kuid enamasti ei saa nad ise rääkida.
Üldiselt venib füüsiline ja vaimne areng, mis avaldub haiguse kulgemisel suureneva alaarengu ja mitmesuguste füüsiliste piirangute kaudu. Lisaks võib Angelmani sündroom põhjustada palju muid sümptomeid. Mõjutatud lastel on sageli epilepsiahooge või nad kannatavad nn mikrotsefaalia, pea, mis on liiga väike.
Täiendavad välised tunnused on väljendunud lõualuu ja lai suu, laiali asetsevad hambad, strabismus ja laiade jalgadega jalutamine väljapoole pööratud. Mõjutatud lapsi võluvad ka vesi, lõhenev paber ja plast ning neil on ebaharilikud söömisharjumused. Tüüpiline on ka halvasti pigmenteerunud nahk koos heledate juuste ja heledate silmavärvidega.
Diagnoos ja kursus
Praeguseks on Angelmani sündroomi diagnoosimiseks teada kaks meetodit. Kolme kuni seitsme aasta vanuselt märkavad lasteneuroloogid EEG väärtusi ebanormaalselt.
Positiivne geneetiline test annab kindluse, kas laps sündis Angelmani sündroomiga. Imikueas ja varases väikelapse eas on Angelmani sündroomi olemasolevad arenguprobleemid vaevalt märgatavad. Diagnoositakse keskmise sagedusega 1: 15 000 kuni 1: 20 000.
Selle eripära kohta võib olla veel mitu juhtumit. Angelmani sündroomi sageli ei diagnoosita. Selle asemel eeldatakse, et see on autism. Nii poistel kui ka tüdrukutel on ühesugune võimalus sündida Angelmani sündroomiga. Angelmani sündroomiga inimeste eeldatav eluiga on normaalne. Varase abi parandamine võib nende iseseisvust märkimisväärselt parandada.
Tüsistused
Angelmani sündroomi puhul on tegemist peamiselt vaimse ja füüsilise arenguga. Asjaomane inimene naerab sageli ilma põhjuseta. See võib paljudele inimestele tunduda tõrjuv ja kummaline, nii et nad isoleerivad neid sotsiaalselt. Lisaks täheldatakse hüperaktiivset ja hüpermotoorset käitumist, nii et vigastuste oht on kõrge.
Angelmani sündroomil on tõenäolisem epilepsiahoog, mis võib põhjustada vigastusi. Lisaks võib tekkida eluohtlik epileptiline staatus. See eriti pikaajaline rünnak võib esineda mitu korda järjest ja seda on raske ravida, mistõttu tuleks viivitamatult kutsuda erakorraline arst ja kavandada intensiivne arstiabi.
Lisaks on patsiendil kilpimine (strabismus). Esimestel aastatel tuleks seda ravida nii kiiresti kui võimalik, kuna koolieas ei saa seda enam tagasi pöörata ja see võib põhjustada halva nägemise (amblüoopia). Pärast operatsiooni on alati oht, et koormus naaseb, mistõttu on vajalik veel üks silmalihaste operatsioon.
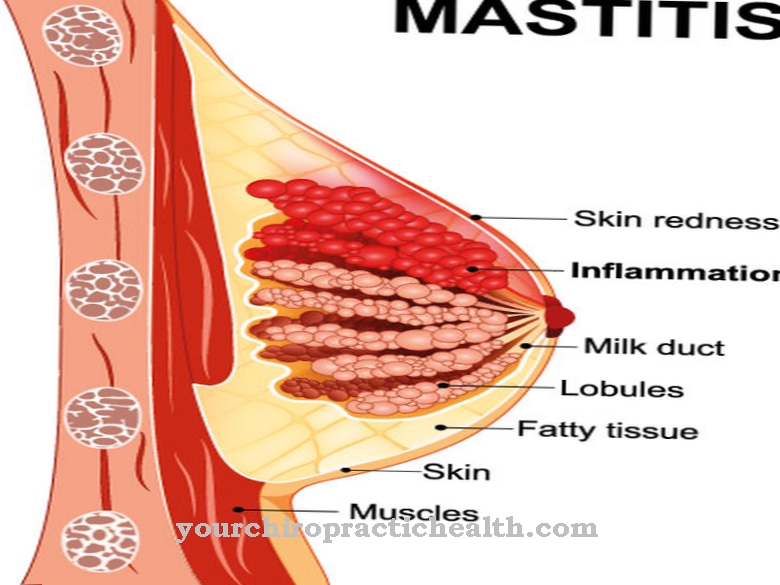
Angelmani sündroomi teine komplikatsioon on selgroo kõverus (skolioos). Aastate jooksul selgroolülide kehad ja nendevahelised kettad kuluvad üha enam, põhjustades tugevat valu. Samuti muutub rindkere väiksemaks, organid on kitsendatud ja nende funktsioonid on piiratud, näiteks süda või kopsud.
Millal peaksite arsti juurde minema?
Angelmani sündroomi kahtluse korral peaksid vanemad rääkima oma lastearsti või lasteneuroloogiga. Selle seisundi kindlakstegemiseks võib kasutada mitmesuguseid füüsilisi sümptomeid ja käitumist. Sellise kliinilise pildi (hilinenud vaimne ja füüsiline areng, liikumis- ja tasakaaluprobleemid jne) ilmnemisel on soovitatav arst.
Eelkõige peaks meditsiiniline hinnang põhinema põhijoonel, väga sagedastel ja alusetutel naeratustel. Sümptomid ilmnevad tavaliselt vanuses 3–7 aastat ja neid saab määratleda infovoldikute ja foorumite abil. Sellegipoolest peaks meditsiiniline diagnoos aset leidma, et kiiresti saaks alustada sobivaid ravimeetmeid.
Ravi peaksid alati olema arutelud psühholoogide ja teiste mõjutatud vanematega. Vanemad peaksid kaaluma ka Angelmani sündroomiga laste spetsiaalsetes vestlusringides osalemist. Mõnel juhul Angelmani sündroomiga tuleb kutsuda erakorraline arst. Siis näiteks siis, kui tekivad krambid või liikumis- ja tasakaaluprobleemide tagajärjel juhtub õnnetus.
Teie piirkonna arstid ja terapeudid
Ravi ja teraapia
Angelmani sündroom pole praegu ravitav. Kuid piisavad meetmed füüsiliste kaebuste raviks on meditsiiniliselt asjakohased. Füsioteraapia sobib lülisamba stabiliseerimiseks, et tasakaalustada skolioosist põhjustatud võimalikke tagajärgi, mis esinevad paljudel Angelmani sündroomiga inimestel.
Epilepsiahoogude vältimiseks võib olla vajalik anda sobivaid ravimeid. Angelmani sündroomiga inimestel võib olla keeleoskus häiritud ja nad ei räägi peaaegu kunagi ise sellest hoolimata, kuid neil on keelt hästi mõista. Professionaalse toega on mõjutatud isikud võimelised õppima alternatiivseid suhtlusvorme, näiteks viipetoega suhtlemist või visuaalset suhtlust.
Terapeutiline ratsutamine võimaldab Angelmani sündroomiga lastel parandada tasakaalutunnet ja seeläbi ohutumalt joosta. Teised terapeutilised abinõud peenmotoorika, iseseisvuse ja kehateadlikkuse parandamiseks on tegevusteraapia, logopeediline ja sensoorse integratsiooni teraapia.
Lisaks on tähelepanelikul hooldamisel, tähelepanu armastamisel ja intensiivsel haridustoel positiivne mõju Angelmani sündroomiga inimestele.
Outlook ja prognoos
Angelmani sündroom mõjutab reeglina väga negatiivselt patsiendi arengut ja võib seda oluliselt edasi lükata. Piiratud pole mitte ainult patsiendi vaimne, vaid ka füüsiline ja motoorne areng, nii et täiskasvanueas on tõsiseid piiranguid ja raskusi. Mitte harvemini sõltuvad kannatanud siis teiste inimeste abist. See põhjustab koordineerimise ja keskendumise häireid. Lugemine ja kirjutamine on tavaliselt keeruline ka neil, keda mõjutab Angelmani sündroom ja esinevad kõnehäired.
Lisaks võivad patsiendid kannatada ka epilepsiahoogude all, mis halvimal juhul võib põhjustada surma. Puudutatud kannatanute käitumine on hüperaktiivne, mille tagajärjel kannatavad vanemad ja sugulased sageli psühholoogiliste kaebuste või depressiooni all.
Selle haiguse otsene põhjuslik ravi ei ole tavaliselt võimalik. Need, kes on kannatanud, sõltuvad kogu elu ravimeetoditest ja teiste inimeste abist. Reeglina ei saa universaalselt ennustada, kas haigus viib ka oodatava eluea lühenemiseni.
ärahoidmine
Angelmani sündroomi ei saa vältida. Geneetiline testimine raseduse ajal võimaldab tuvastada Angelmani sündroomi sündimata lapsel. Sellise protseduuriga kaasneb siiski palju riske ja seda tuleks läbi viia alles pärast hoolikat kaalumist, konsulteerides raviarstiga. Ultraheliuuring ei suuda Angelmani sündroomi kindlaks teha.
Järelhooldus
Angelmani sündroomi korral on järelravi võimalused väga piiratud. Kuna see on pärilik haigus, ei saa seda täielikult ja isegi ainult sümptomaatiliselt ravida. Haigestunud inimene sõltub enamasti elukestvast ravist.
Kui soovite ikkagi lapsi saada, võib läbi viia ka geneetilise nõustamise, et vältida Angelmani sündroomi edasikandumist lastele. Sündroomi ravi toimub tavaliselt füsioteraapia ja erinevate harjutuste abil. Neid saab läbi viia ka oma kodus, mis suurendab oluliselt asjaomase inimese liikuvust.
Enamikul juhtudel on sugulaste ja pereliikmete intensiivsel ja armastaval hooldamisel väga positiivne mõju ka haiguse edasisele kulgemisele. Angelmani sündroomi korral vajab patsient oma igapäevaelus alati tuge ega saa sageli üksi hakkama. Intensiivne tugi võib leevendada ka intellektuaalseid kaebusi. Angelmani sündroom ei piira tavaliselt kannatanute eluiga.
Saate seda ise teha
Kuna haigestunud patsientide füüsilises ja vaimses arengus esinevad viivitused, ei ole nad tavaliselt võimelised võtma omal jõul eneseabimeetmeid. Lisaks ilmneb see geneetiline haigus sünnist alates. Seega saavad väljakutse eeskätt patsiendi pereliikmed.
Angelmanni sündroom on suhteliselt haruldane haigus. Seetõttu peaksid vanemad tagama, et nende lapse eest hoolitseb arst, kellel on selle häirega varasemad kogemused. Kõne areng on enamikul Angelmanni sündroomiga lastest tugevalt aeglustunud. Lapsele suunatud logopeediline tugi ei suuda seda puudujääki korvata, kuid mõnel juhul võib see seda vähemalt vähendada.
Sageli ei suuda mõjutatud isikud siiski oma keskkonnaga keelt kasutades suhelda. Kuid paljud Angelmanni patsiendid oskavad keelt hästi ja on seetõttu võimelised õppima viipekeelt. See võime tuleks mõjutatud isikutele anda võimalikult varakult, kuna suhtlus ümbritsevate inimestega avaldab positiivset mõju intellektuaalsele arengule.
Füsioterapeutilised abinõud aitavad füüsiliste häirete, eriti skolioosi vastu, mis sageli algab puberteedieas. Lisaks saab tegevusteraapia abil edendada tasakaalutunnet ja motoorseid oskusi. Lisaks peaksid kannatanud laste vanemad aegsasti otsima sobivat erikooli, kus juhendatakse ka viipekeeles.





.jpg)